Besiedlung der Ostsee als mariner Extremlebensraum
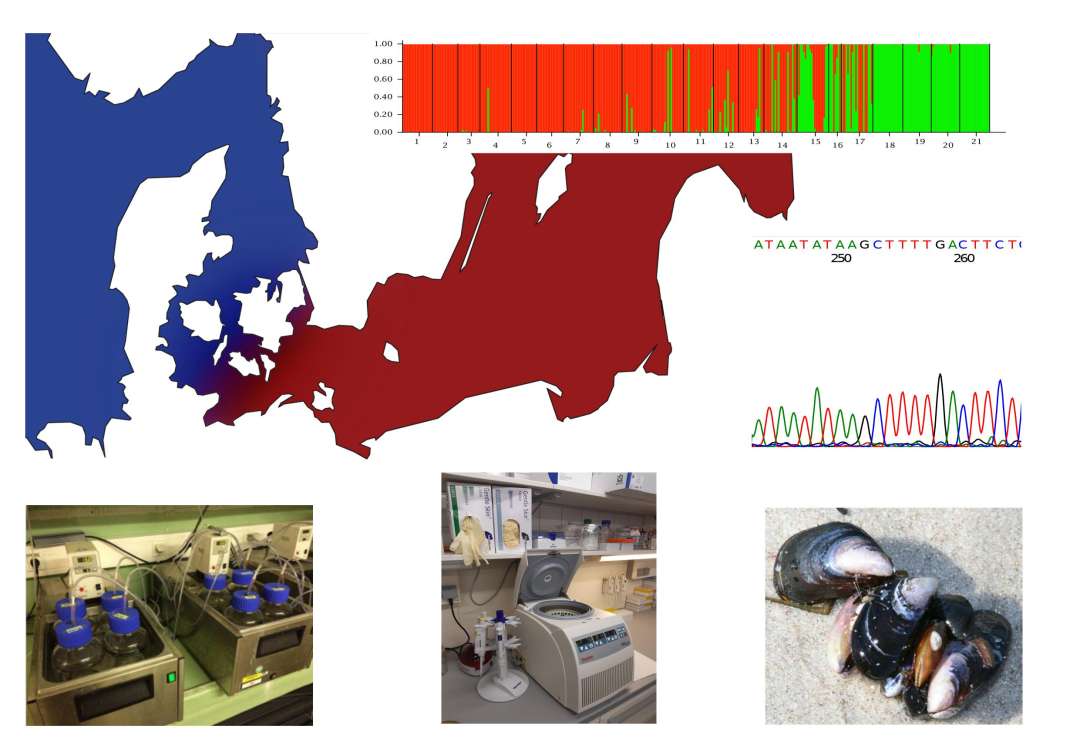
Im Mittelpunkt des Forschungsschwerpunktes steht die Frage, wie Miesmuscheln (Mytilus edulis, M. trossulus, M. galloprovincialis) marine Lebensräume mit geringer Salinität (Brackwasser) besiedeln können. Unter niedriger Salinität stellen Prozesse wie die Osmoregulation oder die Schalenbildung (Biomineralisation) eine physiologische Herausforderung dar. In Fachgebiet Populationsgenetik wird ein integrativer Forschungsansatz verwendet (z.B.: Populationsgenetik, Molekularbiologie, Ökophysiologie), um Miesmuscheln in der Ostsee zu untersuchen, wo sie bis zu 90% der benthischen Biomasse ausmachen. Miesmuscheln kommen in der westlichen Ostsee (Skagerrak/Kattegat; M. edulis; 25-12 practical salinity units (psu)) und der östlichen Ostsee (M. trossulus; 8-4.5 psu) vor. Zum Vergleich: M. edulis lebt in der Nordsee bei ca. 30 psu. Die Entdeckung massiver Hybridisierung (M. edulis x M. trossulus – Hybridschwarm) führte zu der Vermutung, daß der zwischenartliche Genfluß ursächlich für die Fähigkeit sein könnte, um die Brackwasserbedingungen der Ostsee-Gebiete tolerieren zu können. Wir testen diese Hypothese gemeinsam mit alternativen Erklärungen: demographische Prozesse (z.B.: Larvaldrift, Verschiebung der Hybridzone), prä- und post-zygotische reproduktive Barrieren, differentielle Selektion (z.B.: Selektion für Anpassung an lokale Umweltbedingungen) oder neutrale genetische Effekte. Diese Forschung soll nicht nur evolutionäre Prozesse beleuchten, sondern auch Grundlagen schaffen, die Auswirkungen globaler Klimaveränderungen abzuschätzen. Es ist in der Tat so, daß für die Ostsee-Gebiete ein Absinken des Salzgehaltes im Rahmen des Klimawandels vorausgesagt wird. Da Salinität ein entscheidender Umweltfaktor in marinen Umwelten ist, könnte das Auswirkungen auf die Biodiversität der Ostsee (inklusive Verbreitung der Miesmuscheln) haben. Ökosystem-Veränderungen der Ostsee betrifft 9 Staaten und ca. 85 Millionen Menschen in den Anrainergebieten.
Heiko Stuckas
Figure 2: Integrative Forschung (z.B.: Genetik, Genomik, Laborzuchten, Ökophysiologie, Phänotypanalyse) zur Korrelation von genomischen Unterschieden von Ostsee-Miesmuscheln und ihrer Fähigkeit, Brackwassergebiete zu besiedeln. Brackwasser stellt eine besondere Herausforderung für physiologische Prozesse wie Schalenbildung (Biomineralisation) dar.
Wichtige Publikationen zum Forschungsschwerpunkt
- Knöbel L, Nascimento-Schulze JC, Sanders T, Zeus D, Hiebenthal C, Barboza FR, Stuckas H*, Melzner F* (2021): Salinity driven selection and local adaptation in Baltic Sea Mytilid mussels. Frontiers in Marine Science, published open access on 13th of August 2021. (*shared senior authorship)
- Knoebel L, Breusing C, Bayer T, Sharma V, Hiller M, Melzner F, Stuckas H (2020): Comparative de novo assembly and annotation of mantle transcriptomes from the Mytilus edulis species complex (M. edulis, M. galloprovincialis, M. trossulus). Marine Genomics, 51: Article 100700.
- Stuckas H, Knöbel L, Schade H, Breusing C, Hinrichsen HH, Bartel M., Langguth C, Melzner F (2017): Combining hydrodynamic modelling with genetics: Can passive larval drift shape the genetic structure of Baltic Mytilus populations? Molecular Ecology, 26: 2765-2782.
Biogeographie und Populationsstruktur von Tiefseeorganismen
Obwohl sich Tiefseegebiete (Wassertiefen unterhalb von 200m) unter ca. 88% der Meeresoberfläche verbergen, sind diese Bereiche der Erde weitgehend unerforscht. Es gibt große Wissenslücken bezüglich Aspekten zur Biodiversität oder Ökosystemfunktion. Im Fachgebiet Populationsgenetik werden molekulare Forschungsansätze genutzt (z.B.: molekulare Taxonomie, Populationsgenetik), um neue Arten zu identifizieren, deren Verbreitung und Biogeographie zu erforschen und Fragen zur innerartlichen genetischen Variation zu beantworten. Populationsgenetische Ansätze werden ferner auch zur Schätzung von Interaktionen zwischen Populationen genutzt (Konnektivität). Der angewandte Aspekt dieser Forschung gilt der Risikoabschätzung von geplanten Vorhaben zum Tiefseebergbau entlang der mittelozeanischen Rücken (z.B.: Hydrothermalschlote) oder in Tiefseeebenen (Manganknollen).
Wichtige Publikationen zum Forschungsschwerpunkt
- Janssen A*, Stuckas H*, Annemiek Wink, Pedro Martinez Arbizu P (2019): Patterns of genetic connectivity and demograhy in populations of predominant macrofaunal taxa (Polychaeta, Isopoda) in abyssal polymetallic nodule fields: Implications for conservation management. Marine Biodiversity, 49: 2641–2658. (*shared first authorship)
- Dunn DC, Van Dover CL, Etter RJ, Smith CR, Levin LA, T. Morato, Colaço A, Dale AC, Gebruk AV, Gjerde KM, Halpin PN, Howell KL, Johnson D, Perez JAA, Ribeiro MC, Stuckas H, P. Weaver and the SEMPIA Workshop Participants (2018): A strategy for the conservation of biodiversity on mid-ocean ridges from deep-sea mining. Science Advances, 4(2): eaar4313.
- Gollner G*, Stuckas H*, Kihara TC, Kodami S, Martinez Arbizu P (2016): Mitochondrial DNA analyses indicate high diversity, expansive population growth and high genetic connectivity of vent copepods (Dirivultidae) across different oceans. Plos One, 11(10): e0163776. (* shared co-authorship)
Populationsstruktur und Hybridisierung
Dieser Forschungsschwerpunkt zielt auf die Erforschung allgemeiner Prinzipien ab, die zur Ausbildung von Unterschieden zwischen Populationen und zur Hybridisierung von Arten führen. In verschiedenen Kooperationsprojekten arbeiten wir mit verschiedenen Taxa (z.B.: Schildkröten, Sperlingsvögel, Säugetiere). Derartigen Projekten geht oft ein allgemeines Monitoring voraus, was seitens des Fachgebietes Populationsgenetik mit einer Expertise im genetischen Barcoding begleitet wird (z.B.: COI-Barcoding mittels minimal invasiv gesammelter Proben wie Säugetierkot). Die Forschung steht oft im Kontext von Naturschutzgenetik. Genetisches Monitoring in verschiedenen Taxa zeigt aber auch, wie häufig zwischenartliche Hybridisierung vorkommt und hat zu Forschungsarbeiten über die Struktur und Funktionsweise von Hybridzonen von Schildkröten- und Vogelarten geführt. Bezugnehmend auf unsere Forschung an den hybridisierenden Ostsee-Miesmuscheln zielen diese Aktivitäten auch auf ein allgemeines Verständnis der Bedeutung von Hybridisierung in der Evolution ab (z.B., Speziation, „speciation in reverse“, Vermischung von Gen-Pools).

Heiko Stuckas

Heiko Stuckas

Heiko Stuckas
Figure 3: Im Fachgebiet Populationsgenetik werden Kooperationsprojekte in den Bereichen Biomonitoring, Speziation, Hybridisierung und Naturschutzgenetik an verschiedenen Taxa durchgeführt.
Wichtige Publikationen zum Forschungsschwerpunkt
- Wolfgramm H, Martens J, Töpfer T, Vamberger M, Pathak A, Stuckas H, Päckert M (2021): Asymmetric allelic introgression across a hybrid zone of the coal tit (Periparus ater) in the central Himalayas. Ecology and Evolution, 11: 17332-17351.
- Päckert M, Bekacem, AA, Wolfgramm H, Gast O, Canal D, Giacalone G, Lo Valvo M, Vamberger M, Wink M, Martens J, Stuckas H (2019): Genetic admixture despite ecological segregation in a North African sparrow hybrid zone (Aves, Passeriformes, Passer domesticus x Passer hispaniolensis). Ecology and Evolution, 9: 12710 – 12726.
- Vamberger M. Stuckas H, Vargas-Ramírez M, Kehlmaier C, Ayaz D, Aloufi AA, Lymberakis P, Široký P, Fritz U (2017): Unexpected hybridization patterns in Near Eastern terrapins (Mauremys caspica, M. rivulata) indicate ancient gene flow across the Fertile Crescent. Zoologica Scripta, 46: 401-413.
Erforschung genomischer Ursachen zwischenartlicher Unterschiede
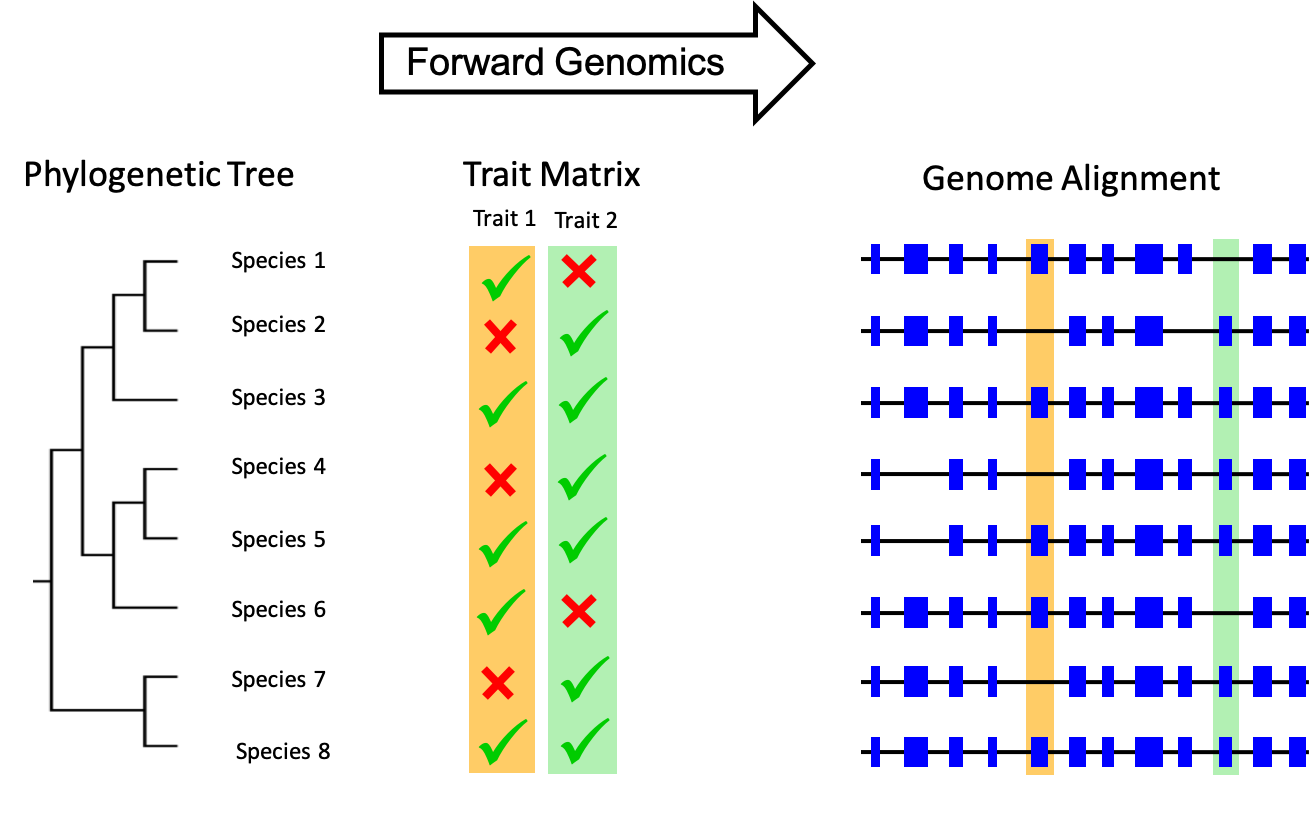
Die phänotypische Vielfalt von Tieren ist enorm und auch einzelne Taxa wie beispielsweise die Säugetiere zeigen ganz unterschiedliche Merkmale und Baupläne. Es stellt sich deshalb die grundsätzliche Frage, auf welche Art und Weise sich Genome in der Evolution verändert haben, um diese phänotypische Variationsbreite hervorzubringen. In einem Pilotprojekt unter maßgeblicher Mitwirkung des Fachgebietes Populationsgenetik wurde in den vergangenen Jahren ein neuer Forschungsansatz angewendet, um genomische Ursachen unterschiedlicher Säugetierphänotypen zu erforschen (Leitung eines Forschungskonsortiums aus 9 Einrichtungen im Rahmen des Leibniz-Wettbewerbs 2016: „Identifying the genomic loci underlying mammalian phenotypic variability using Forward Genomics with Semantic Phenotypes”). In diesem Pilotprojekt wurde Forward Genomics als neues Konzept angewandt. Dabei werden Genomalignments mit Phänotyp-Matrices verglichen, um Phänotyp assoziierte Genverluste zu identifizieren. So wurde beispielsweise das Fehlen eines Rezeptors (RXFP2) mit dem Fehlen des Hodenabstiegs bei Elefanten und deren Verwandte (Säugetiertaxon: Afrotheria) in Zusammenhang gebracht.
Neben Herausforderungen in der vergleichenden Genomik hängt dieser Forschungsansatz maßgeblich vom Vorhandensein von Phänotyp-Informationen ab. Unsere Forschungsarbeiten zeigten, daß es hier immer noch große Wissenslücken gibt! Ferner bedarf es einer allgemein zugänglichen Dokumentation der Merkmale (z.B.: distinkte Kategorien wie Vorhandensein vs. Fehlen) in einem computerlesbaren Format (z.B.: NEXUS-Format). Um diesen Bedarf zu bedienen, engagiert sich das Fachgebiet in einer neu etablierten Forschungsplattform: MaTrics (Mammalian Traits for comparative genomics; https://www.senckenberg.de/de/institute/senckenberg-naturhistorische-sammlungen-dresden/museum-fuer-tierkunde/dd-sekt-mammalogie/matrics-consortium/).
Derzeitige Projekte konzentrieren sich beispielsweise auf anatomische Merkmale (z.B.: Gebiß) oder Ernährung (z.B.: Herbivorie, Carnivorie, Omnivorie). Diese Arbeiten vertiefen nicht nur das Verständnis von Evolution, sondern haben auch Bedeutung für biomedizinische Fragestellungen (Phylomedizin). In der Zukunft soll dieser Forschungsansatz auch auf Nicht-Säugetiere angewandt werden.
Heiko Stuckas

Heiko Stuckas
Figure 4: Forward Genomics ist ein Forschungsansatz, mit dem beispielsweise genomische Ursachen phänotypischer Variation von Säugetieren analysiert werden können.
Wichtige Publikationen zum Forschungsschwerpunkt
- Wagner F, Ruf I, Lehmann T, Hofmann R, Ortmann S, Schiffmann C, Hiller M, Stefen C, Stuckas H (2022): Reconstruction of evolutionary changes in fat and toxin consumption reveals associations with gene losses in mammals: a case study for the lipase inhibitor PNLIPRP1 and the xenobiotic receptor NR1I3. Journal of Evolutionary Biology, 35: 225-239.
- Stefen C, Wagner F, Aszatlos M, Giere P, Grobe P, Hiller M, Hofmann R, Jähde M, Lächele U, Lehmann T, Ortmann S, Peters B, Ruf I, Schiffmann C, Their N, Unterhitzenberger G, Vogt L, Rudolf M, Wehner P, Stuckas H (2021): Phenotyping in the era of genomics: MaTrics – a digital character matrix to document mammalian phenotypic traits. Mammalian Biology, published open access on 7th of December 2021.
- Sharma V, Lehmann T, Stuckas H, Hiller M (2018): Loss of the gubernaculum-inducing INSL3 and RXFP2 genes in afrotheria shows that testicular descent is the ancestral condition in placental mammals. Plos Biology, 16(6): e2005293.